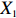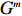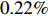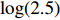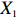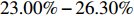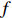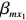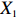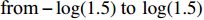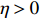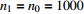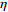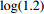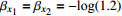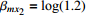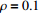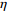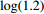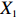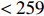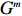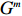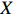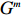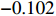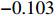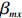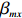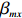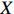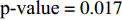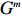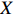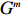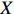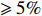Yanan Zhao is currently a PhD student under the tutelage of Prof. Hong Zhang at the University of Science and Technology of China. Her research interests focus on empirical Bayes procedures and statistical genetics
Hong Zhang is a Full Professor with the University of Science and Technology of China (USTC). He received his Bachalor’s degree in Mathematics and Ph.D. degree in Statistics from USTC in 1997 and 2003, respectively. His major reseach interests include statistical genetics, causal inference, and machine learning
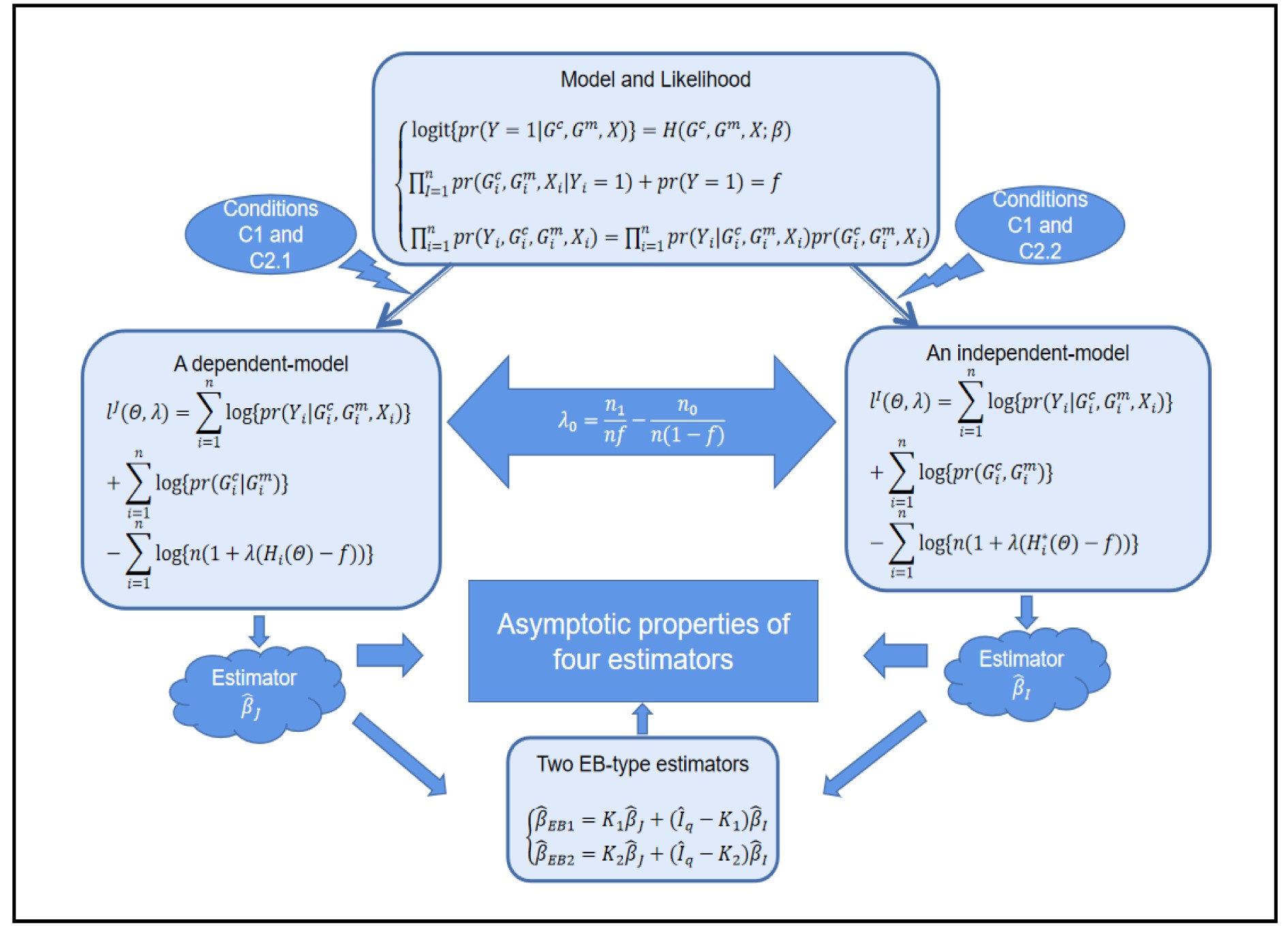
Case-control mother-child pair data are often used to investigate the effects of maternal and child genetic variants and environmental risk factors on obstetric and early life phenotypes. Retrospective likelihood can fully utilize available information such as Mendelian inheritance and conditional independence between maternal environmental risk factors (covariates) and children’s genotype given maternal genotype, thus effectively improving statistical inference. Such a method is robust to some extent if no relationship assumption is imposed between the maternal genotype and covariates. Statistical efficiency can be considerably improved by assuming independence between maternal genotype and covariates, but false-positive findings would be inflated if the independence assumption was violated. In this study, two empirical Bayes (EB) estimators are derived by appropriately weighting the above retrospective-likelihood-based estimators, which intuitively balance the statistical efficiency and robustness. The asymptotic normality of the two EB estimators is established, which can be used to construct confidence intervals and association tests of genetic effects and gene-environment interactions. Simulations and real-data analyses are conducted to demonstrate the performance of our new method.
Graphical Abstract
Based on a dependent-model estimator and an independent-model estimator, we obtaintwo Bayes-type estimators that balance robustness and efficiency.
Abstract
Case-control mother-child pair data are often used to investigate the effects of maternal and child genetic variants and environmental risk factors on obstetric and early life phenotypes. Retrospective likelihood can fully utilize available information such as Mendelian inheritance and conditional independence between maternal environmental risk factors (covariates) and children’s genotype given maternal genotype, thus effectively improving statistical inference. Such a method is robust to some extent if no relationship assumption is imposed between the maternal genotype and covariates. Statistical efficiency can be considerably improved by assuming independence between maternal genotype and covariates, but false-positive findings would be inflated if the independence assumption was violated. In this study, two empirical Bayes (EB) estimators are derived by appropriately weighting the above retrospective-likelihood-based estimators, which intuitively balance the statistical efficiency and robustness. The asymptotic normality of the two EB estimators is established, which can be used to construct confidence intervals and association tests of genetic effects and gene-environment interactions. Simulations and real-data analyses are conducted to demonstrate the performance of our new method.
Public Summary
Retrospective likelihood method is employed to improve statistical efficiency by fullyutilizing available information.
An efficient estimator and a robust estimator are combined to construct twonovel empirical Bayes-type estimators using empirical the Bayes method.
We establish the asymptotic properties of the proposed estimators.
It is well known that most obstetric and early-life diseases or phenotypes have a multifactorial etiology involving genetic factors, environmental exposures, and interactions between them[1–4]. For example, low maternal body mass index and genes of the mother and child were found to be associated with the risk of preterm delivery[5, 6]. Moreover, maternal COL24A1 variants were shown to have significant genome-wide interactions with maternal pre-pregnancy overweight/ obesity on preterm birth risk using genetic data of 1733 African-American women from the Boston Birth Cohort[7]. Identifying potential genetic risk factors is important for understanding their biological effects and for developing public health strategies for prevention. A popular design for the integrative investigation of the effects affecting obstetrical and early life phenotypes is to collect risk factor information from children and their mothers[1, 8, 9]. In this study, the outcome of interest was a dichotomous phenotype of the mother or child, and cases and controls were ascertained based on this phenotype. This is the so-called mother-child case-control design[10, 11].
The standard prospective logistic regression method[12] is adopted widely in case-control studies. However, this method is not efficient because it does not utilize information such as Mendelian inheritance between parental and child genotypes. Alternative methods have been developed to assess the genetic effects on both children and mothers. Shi et al.[13] and Chen et al.[8] proposed the log-linear method and retrospective method for fitting logistic regression models, respectively, and improved the statistical inference efficiency by incorporating constraints on the genotype distribution, such as Mendelian inheritance, random mating, and Hardy-Weinberg equilibrium (HWE) in the population of interest. Chen et al.[10] extended the retrospective likelihood method to allow assessment of environmental effects and gene-environment interactions through a semiparametric maximum likelihood estimation (MLE) method, and improved statistical inference efficiency by incorporating an additional constraint that children’s genotype is conditionally independent of maternal environmental risk factors given maternal genotype.
Using the retrospective likelihood method to analyze case-control mother-child pair data can result in a noticeable efficiency gain in estimating odds ratio (OR) parameters under the model assumptions of HWE and maternal gene-environment independence in the population of interest; however, it would produce serious bias if the independence assumption or HWE is violated. Chen et al.[10] alleviated the bias concern by allowing the conditional distribution of environmental risk factors for a given maternal genotype to be nonparametric, but this strategy is not sufficiently efficient. Alternatively, Zhang et al.[14] proposed a modified profile likelihood estimation method by relating the maternal genotype to environmental risk factors through a novel “double-additive” logistic regression (daLOG) model, which is computationally robust but not sufficiently robust because estimation bias may be introduced when the daLOG model is seriously violated.
Two novel estimators are proposed in this study: the weighted averages of an efficient estimator and a robust estimator. The second estimator is robust in that it is nearly unbiased, and the corresponding significance test has Type I error controlled around the nominal level. Both estimators are closely related to the modified profile MLE of Zhang et al.[14]. The efficient one imposes the assumptions of HWE and maternal gene-environment independence (referred to as “independent-model” estimator hereafter), which is biased if at least one of the assumptions is violated. In contrast, the robust one does not require the two assumptions, so it is relatively robust but might not be efficient (refer to as “dependent-model” estimator hereafter). Our weighting method was motivated by an empirical Bayes-type (EB-type) shrinkage estimator for estimating the effects of haplotypes and haplotype-environmental interactions with standard case-control data[15]. Here, such shrinkage technique is extended to assess OR parameters associated with maternal genotype and environmental risk factors using case-control mother-child genotype data. Briefly, the robust “dependent-model” estimator is shrunk toward the more efficient but possibly biased “independent-model” estimator. The magnitude of “shrinkage” is data-dependent, which tends to be large if and only if HWE and maternal gene-environmental independence are satisfied.
The remainder of this paper is organized as follows. In Section 2, two MLEs are derived based on dependent and independent models. Subsequently, two EB-type estimators are constructed based on the two MLEs, and their asymptotic properties are established. In Sections 3 and 4, the desired finite-sample performance of the proposed EB-type estimators is demonstrated through extensive simulations and real-data applications. Concluding remarks are provided in Section 5.
2.
Materials and methods
2.1
Model, likelihood, and assumptions
Let Y denote the dichotomous disease status of either mother (e.g., pre-eclampsia) or child (e.g., low birth weight), that is, Y=0 for a control pair and Y=1 for a case pair, (Gm,Gc) denote the genotypes of a mother-child pair at a target genetic locus, and X denotes a vector of p maternal environmental risk factors. Y and (Gc,Gm,X) are related using the following logistic penetrance model:
logit{pr(Y=1|Gc,Gm,X)}=H(Gc,Gm,X;β)
(1)
where logit(t)=log(t/(1−t)) is the logit function, H(⋅) is an arbitrarily specified function, and β is the q-vector of the regression parameters. Gc and Gm can be coded according to the mode of inheritance. The code set for a specific mode of inheritance is denoted as M. For the additive mode of inheritance, the genotype is coded as the number of minor alleles (M={0,1,2}); for the dominant mode of inheritance, the genotype is coded as 1 if at least one of the minor alleles is present and 0 otherwise (M={0,1}); for the recessive mode of inheritance, the genotype is coded as 1 if both alleles are minor alleles and 0 otherwise (M={0,1}).
Suppose that (Gc,Gm,X) is collected from the n0 control pairs (Y=0) and n1 case pairs (Y=1). Let n=n0+n1. The retrospective likelihood for case-control mother-child data is
n∏i=1pr(Gci,Gmi,Xi|Yi).
Assume that the phenotype prevalence pr(Y=1) is known a priori to be f or that it can be estimated using extra data. According to the Bayesian theorem, under constraint
pr(Y=1)=f
(2)
maximizing the likelihood function above is equivalent to maximizing:
In this study, all the statistical inference procedures were based on the likelihood function (3). Under Mendelian inheritance and random mating, the joint probability pr(Gci,Gmi,Xi) in (3) can be further decomposed according to the two distributional assumptions on (Gc,Gm,X) (C1 and C2.1/C2.2).
C1. The child genotype is independent of maternal environmental risk factors conditional on the maternal genotype, i.e., pr(Gci|Gmi,Xi)=pr(Gci|Gmi).
C2.1. The joint distribution (Gmi,Xi) is completely unspecified (dependent model).
C2.2. Gmi is independent of Xi (independent model).
Maternal environmental risk factors are generally maternal-related characteristics; therefore, they should be independent of the child genotype given the maternal genotype. Thus, C1 is a reasonable assumption in studies of gene-environment associations with obstetrical/maternal phenotypes using case control mother-child pair data. In contrast, maternal genotype and environmental factors can be correlated or uncorrelated, corresponding to Conditions C2.1 and C2.2, respectively.
In the following subsections, we modify the estimator proposed by Zhang et al.[14] to obtain two estimators of the regression parameter vector β=(β0,βT1)T under Conditions C1 and C2.1/C2.2. The first estimator based on C2.1 is robust but inefficient, and the second estimator is efficient but can be biased if C2.2 is violated. We then extend the EB theory of Chen et al.[15] to derive a shrinkage estimator that can adaptively balance bias and efficiency.
2.2
A dependent-model based estimator
Under Condition C1, the joint probability pr(Gc,Gm,X) can be written as
pr(Gc,Gm,X)=pr(Gc|Gm)pr(Gm,X).
If Mendelian inheritance and random mating hold in the population of interest, the conditional distribution pr(Gc|Gm) is a function of MAF θ and fixation index ρ (a parameter measuring deviation from HWE); refer to Ref. [14, Table 1] for the conditional distributions under various modes of inheritance. These distributional constraints are incorporated into the likelihood function (3): Under C2.1, the empirical likelihood method[16] can be adopted by introducing the probability mass πi for pr(Gmi,Xi) that satisfies the constraint
n∑i=1πi=1
(4)
Denote π=(π1,…,πn). The empirical log-likelihood function can be written as:
As shown in Supporting Information Appendix S1, under constraints (2) and (4), the profile likelihood function of Θ=(θ,ρ,βT)T can be obtained using the Lagrange multiplier method, which takes the form:
which is exactly the “score” equation derived from profile likelihood lJ with respect to λ. Then, Θ and λ can be estimated by solving Eq. (7) and ∂lJ(Θ,λ)/∂Θ=0,
As show in Ref. [17], it is computationally unstable to estimate Θ and λ using the above method as the solution is a saddle point of lJ. The profile-likelihood function (6) can be modified to resolve this numerical problem. Specifically, λ in Eq. (6) can be replaced by its limiting value:[17]
λ0=n1nf−n0n(1−f)
(8)
The validity of this modification is as follows. Let Θ0 be the true value of Θ. The “true” value λ0 is the solution to the equations (refer to Supporting Information Appendix S2 for the detailed proof):
E[∂lJ(Θ,λ)∂Θ]|Θ=Θ0,λ=λ0=0 and E[∂lJ(Θ,λ)∂λ]|Θ=Θ0,λ=λ0=0
(9)
As a result, the so-called dependent-model estimator of Θ can be obtained by solving
∂lJ(Θ,λ0)∂Θ=0
(10)
Asymptotic properties of the dependent-model estimator can be established under some regularity conditions[18]. First, with a probability tending to one as n→∞, there exists a solution ˆΘJ to Eq. (10), which is consistent with Θ0. Second, ˆΘJ is asymptotically normally distributed (refer to Supporting Information Appendix S3 for a detailed derivation).
Obviously, IJ(Θ0) can be consistently estimated by the empirical Fisher information matrix, that is,
ˆIJ(ˆΘJ):=−1n∂2lJ(Θ,λ0)∂Θ∂ΘT|Θ=ˆΘJ
(12)
Among n selected subjects, without loss of generality, assume that the first n0 subjects are controls (i.e., Yi=0 for i=1,⋯,n0) and the remaining n1=n−n0 subjects are cases (i.e., Yi=1 for i=n0+1,⋯,n=n0+n1). Then, ΣJ(Θ0) can be estimated consistently using
where a⊗2=aaT for any vector a. Therefore, the limiting variance-covariance matrix of √nˆΘJ can be consistently estimated using ˆI−1J(ˆΘJ)ˆΣJ(ˆΘJ)ˆI−1J(ˆΘJ). Consequently, significance tests of the genetic effects can be constructed.
2.3
An independent-model based estimator
Now, we consider obtaining an independent-model estimator of Θ under Conditions C1 and C2.2. In this case, the joint probability pr(Gc,Gm,X) can be written as pr(Gc,Gm)pr(X). Under Mendelian inheritance and random mating, pr(Gc,Gm) is a function of θ or (θ, ρ) depending on whether HWE holds or not. The empirical likelihood method was adopted, as in the dependent model. Here, the distribution of X, instead of the joint distribution of (Gm,X), is allowed to be nonparametric. Let μi denote the probability mass for pr(Xi) that satisfies
n∑i=1μi=1
(14)
Denote μ=(μ1,⋯,μn). Then, the corresponding empirical log-likelihood function is
Similar to the derivation of lJ(Θ,λ), under constraints (2) and (14), the profile likelihood function corresponding to Eq. (15) takes the form (refer to Supporting Information Appendix S1 for a detailed derivation):
The MLEs of Θ and λ∗ can then be obtained by solving the “score” equations
∂lI(Θ,λ∗)∂λ∗=0and∂lI(Θ,λ∗)∂Θ=0
(17)
Again, λ∗ can be replaced by λ0 defined by Eq. (8). Similarly, λ0 is the exact solution to the following equations:
E[∂lI(Θ,λ)∂Θ]|Θ=Θ0,λ=λ0=0 and E[∂lI(Θ,λ)∂λ]|Θ=Θ0,λ=λ0=0
(18)
As a result, the so-called independent-model estimator of Θ can be obtained by solving
∂lI(Θ,λ0)∂Θ=0
(19)
As Supporting Information Appendix S3 shows, the independent model estimator, denoted by ˆΘI, is asymptotically normal under some regularity conditions:
√n(ˆΘI−Θ0)D⟶N{0,I−1I(Θ0)ΣI(Θ0)I−1I(Θ0)}
(20)
where II(Θ0) and ΣI(Θ0) are the analogs of IJ(Θ0) and ΣJ(Θ0), respectively. As in Eqs. (12) and (13), II(Θ0) and ΣI(Θ0) can be estimated consistently.
2.4
Two empirical Bayes-type shrinkage estimators
Compared with the dependent-model estimator, the independent-model estimator is more efficient, but serious bias could be produced if environmental risk factors are correlated with maternal genotype. Chen et al.[15] were fully aware of the two possibly conflicting goals of improving efficiency and maintaining robustness when estimating the effects of haplotypes and haplotype-environmental interactions with standard case-control data and thus proposed an EB-type shrinkage estimator to balance efficiency and robustness in a data-adaptive fashion. Here, we adopt this “shrinkage” estimation technique to balance the estimation robustness and efficiency of the effects of maternal and child genetic variants and environmental risk factors in case-control mother-child studies.
Let ˆβI and ˆβJ denote all genetic and environmental related effect estimates in the independent-model and dependent-model estimators, respectively. Intuitively, a weighted estimator has the form
ˆβW=KˆβJ+(Iq−K)ˆβI=ˆβI+K(ˆβJ−ˆβI)
(21)
where Iq denotes an identity matrix of size q=dim(β), K is a weight factor that favors ˆβI or ˆβJ depending on the bias of independent-model estimator. Obviously, K should have the property that it favors the robust dependent-model estimator if the independent-model estimator is biased, and vice versa. In the following, we derive the optimal weight matrix K based on the EB theory.
Throughout this study, we assume that the dependent model described in Section 2.2 is true. We denote the true value of β as βJ. Let βI be the value of β that minimizes the Kullback-Leibler discrepancy between dependent and independent models. Then, ˆβI converges to βI under certain regularity conditions. The prior distribution of βJ is assumed to be a multivariate normal distribution with expectation βI and variance-covariance matrix A, where A is a q×q positive definite matrix. In view of the asymptotic normality of ˆβJ, conditional on the value of βJ, ˆβJ approximately follows a multivariate normal distribution with mean vector βJ and variance-covariance matrix V, where V is a q×q positive definite matrix. Consequently, the posterior expectation of βJ is given by
A(A+V)−1ˆβJ+V(A+V)−1βI
(22)
where βI is estimated using ˆβI. The appropriate estimators of A and V are discussed as follows. Obviously, ψ:=βJ−βI follows a multivariate normal distribution with a zero mean vector and variance-covariance matrix A. The prior hyperparameter A can be conservatively estimated using ˆψˆψT, where ˆψ=ˆβJ−ˆβI[15, 19]. An estimator of V, denoted by ˆV, can be derived from the variance-covariance matrix of ˆΘJ described in Section 2.2. The resulting EB-type shrinkage estimator can be expressed as
ˆβEB1=K1ˆβJ+(Iq−K1)ˆβI
(23)
where the shrinkage factor, K1, is
K1=ˆψˆψT(ˆψˆψT+ˆV)−1.
As in Ref. [15], another shrinkage factor K2 alternative to K1 is considered, which is a diagonal matrix with the ith diagonal element ki=ˆψ2i/(ˆvi+ˆψ2i):
K2=diag(ˆψ21/(ˆψ21+ˆv1),⋯,ˆψ2q/(ˆψ2q+ˆvq)),
where ˆvi and ^ψi are the ith diagonal elements of ˆV and ˆψ, respectively. This results in an alternative EB estimator:
ˆβEB2=K2ˆβJ+(Iq−K2)ˆβI
(24)
If the observed data support the independence of environmental risk factors and maternal genotype, then ˆψ≈0q such that both ˆβEB1 and ˆβEB2 approximates ˆβI. If the observed data support the correlation between environmental risk factors and maternal genotype, then ˆψˆψT tends to be a non-zero q-dimensional matrix, which increases with the degree of deviation of the independent model; thus, ˆβEB1 and ˆβEB2 would tend to ˆβJ.
In the following, the variance-covariance matrix of ˆβEB1 and ˆβEB2 is derived by applying the delta method. Note that they are functions of (ˆβI,ˆβJ). We first derive the asymptotic distribution of (ˆβI,ˆβJ). Based on the asymptotic expressions of ˆΘJ and ˆΘI derived in Sections 2.2 and 2.3, we obtain the following asymptotic normality:
Here, 0 is a q×2 matrix of zeros, and IJ(Θ0), II(Θ0), ˆIJ(ˆΘJ), and ˆII(ˆΘI) are given in Sections 2.2 and 2.3. Furthermore, S0I(Θ) and S0J(Θ) are the partial derivative vectors of lI(Θ,λ0) and lJ(Θ,λ0), respectively, with respect to Θ.
According to the delta method, the asymptotic variance-covariance matrix of ˆβEB1 and ˆβEB2 can be consistently estimated by (See Supporting Information Appendix S4)
Note that ˆG1 and ˆG2 are the partial derivative matrices of ˆβEB1 and ˆβEB2 with respect to (ˆβTI,ˆβTJ)T.
3.
Simulation studies
The finite sample performance of the proposed method was evaluated through extensive simulations. First, we evaluated the estimation performance for the two EB-type estimators (23) and (24). The powers and Type I error rates of the corresponding significance tests were then examined.
3.1
Simulation setups
Several parameter combinations were considered, as described in the following subsections. For each parameter combination, genotype data (Gm,Gc) were generated for a population of 1×107 mother-child pairs, and two covariates were considered: a continuous covariate X1 and a discrete covariate X2, where X1 was generated according to a linear model and could be either independent of or correlated with maternal genotype, and X2 was sampled from the binomial distribution B(1,0.5) and was independent of both maternal genotype and X1. Given each vector (Gm,Gc,X1,X2), a binary disease outcome Y was generated according to the logistic regression model:
For each parameter combination, 1000 datasets, each consisting of n1 case pairs and n0 control pairs, were sampled from the population.
In all simulations, both Gm and Gc were coded as minor allele counts, so that the additive mode of inheritance was adopted in the penetrance model (1). Note that pr(Gc|Gm) does not depend on the fixation index in this situation (refer to Ref. [14, Table 1]). Hereafter, let Dep represent the dependent-model-based method that does not depend on the HWE assumption. Let IndHWE and IndHWD represent the independent-model-based methods with and without the HWE assumption, respectively. Here, the HWD denotes Hardy-Weinberg disequilibrium (ρ>0). Let θ be fixed at 0.2, and let ρ be fixed at 0 for HWE or 0.1 HWD. The covariate X1 was generated according to the following linear model:
X1=η×[Gm−E(Gm)]+e,
where e was independent of Gm, and follows a standard normal distribution. Note that η characterizes the correlation strength between X1 and Gm, and a zero value of η indicates independence between X1 and Gm and vice versa. In addition, the prevalence of Y was fixed at 0.03 and assumed to be known in the relevant methods.
The value of η was set to 0, log(1.5), or log(2.5) for various correlation strengths between X1 and Gm. The odds ratio parameters were set to βc=log(1.8), βm=log(2), βx1=βx2=−log(1.2), βcx1=βcx2=−log(1.5) and βmx1=βmx2=log(1.5). We considered various sample-size combinations with equal or unequal numbers of case and control pairs. In the equal situation, the common number was set to 150, 300, 600, or 1000; in the unequal situation, the ratio of case pairs to control pairs was set to be 1:2, and the number of case pairs was set to be 100, 200, 400, or 700.
In significance tests, to illustrate the impact of the correlation between maternal genotype and environmental risk factors, the value of η was set to range between 0 and log(2.5), and the values of βc, βx, and βcx were fixed as above, βm was set as log(1.3), βmx2 was set to log(1.2), and the value of βmx1 ranged between −log(1.5) and log(1.5). The significance level was fixed at 5%, and the sample size was n1=n0=150, 300, or 1000.
3.2
Estimation results
Five considered methods, namely IndHWE, IndHWD, Dep, EB1, and EB2 were applied to the generated data. To compare the estimation efficiencies of two estimators, we introduced the so-called efficiency gain of estimator A against estimator B as
1−MSE(A)MSE(B),
where MSE(A) and MSE(B) are the mean squared errors of Methods A and B, respectively. The estimation results are summarized in Tables 1–3 and the Supporting Information Tables S1–S9. The efficiency gains with equal numbers of case and control pairs are presented in Supporting Information Tables S10–S15.
Table
1.
MSE and bias (in parenthesis) of various estimators when case and control pairs are equal and both the independence assumption (η=0) and HWE (ρ=0) hold.
Table
2.
MSE and bias (in parenthesis) of various estimators when case and control pairs are equal and the independence assumption is violated (η=log(1.5)) but HWE holds (ρ=0).
Table
3.
MSE and bias (in parenthesis) of various estimators when case and control pairs are equal and the independence assumption is violated (η=log(2.5)) but HWE holds (ρ=0).
As shown in Tables 1 and S4, all five considered methods are essentially unbiased for all sample size combinations when both HWE and independence assumptions are satisfied. According to MSEs, IndHWE is more efficient than EB1 and EB2, and the latter two methods are more efficient than Dep. For example, when n1=n0=150, the efficiency gains of EB1 and EB2 against Dep were approximately 0.95%−24.29% and 2.16%−37.98%, respectively. IndHWE appears to be much more efficient than IndHWD with efficiency gains ranging between 9.04% and 44.01% for n1=n0=150, although IndHWD incorporates only one more parameter ρ compared with IndHWE (Supporting Information Table S10). When the independence assumption holds but HWE is violated, IndHWE could produce considerable biases on maternal genetic effects but not on the other effects in our simulation situation, so that IndHWD outperforms IndHWE in estimating maternal genetic effects (Supporting Information Tables S1, S7, and S13). However, the estimation results of the remaining four methods appeared to be unaffected by the violation of HWE.
When HWE holds, but the Gm-X1 independence assumption is violated (η=log(1.5)), both IndHWE and IndHWD could be seriously biased, especially for main effect of X1 and the interaction effect between and Gm and X1 (Tables 2 and S5). As expected, Dep, which does not require HWE or independence assumptions, is inherently unbiased. Correspondingly, the MSEs of the above two effects were generally much larger for IndHWE than for Dep. Furthermore, EB1 and EB2 can effectively balance the bias and efficiency. The efficiency gains of EB1 and EB2 against IndHWE are considerable for the above two effects. For example, when n1=n0=150, the efficiency gains of EB1 and EB2 against IndHWE for the two effects are approximately 29.67%−40.04% and 68.67%−73.91%, respectively. For the other effects, EB1 and EB2 are less efficient than IndHWE but more efficient than Dep, with efficiency gains against Dep ranging between 0.22% and 10.56% for EB1 and 0.86% and 33.52% for EB2 when the sample size is 150 (Supporting Information Table S11). In addition, EB1 and EB2 are slightly more efficient than IndHWD in the simulation situations. When both HWE and the independence assumption are violated, both IndHWE and IndHWD are biased, and their performances are poorer than those of the other three methods (Supporting Information Tables S2, S8, and S14).
With a larger η of log(2.5), IndHWE and IndHWD are more biased toward the maternal effect, X1 environmental effect, and Gm-X1 interaction (Tables 3 and S6), in contrast with the results for η=log(1.5) (Tables 2 and S5). Consequently, the efficiency gains of EB1 and EB2 against IndHWE and IndHWD increased. For example, with a sample size of 150, the efficiency gains of EB1 and EB2 against IndHWE for the three effects increased to 23.00%−26.30%, 64.75%−72.99%, and 91.51%−92.62%, respectively (Supporting Information Table S12). When HWE was violated, the efficiency gains of EB1, EB2, and Dep against IndHWE and IndHWD were even greater (Supporting Information Tables S3, S9, and S15).
In summary, IndHWE is the preferred method when the assumptions of HWE and maternal gene-environment independence hold. However, when any one of the two assumptions is violated, serious estimation biases can be produced for maternal genetic effects, environmental effects, and maternal gene-environment interactions. Dep is the most robust with respect to the two assumptions, but it is much less efficient than IndHWE, and its performance is much poorer than IndHWE in terms of MSE when independence is satisfied. In contrast, EB1 and EB2 can balance estimation bias and efficiency by appropriately weighting IndHWE and Dep. In the small-sized situation, EB1 is generally more unbiased than EB2, although the former is slightly less efficient. Consequently, EB1 may be preferable to EB2 with a small sample size; however, EB2 may be preferable to EB1 with a moderate or large sample size.
The five considered methods require specification of the prevalence f. Therefore, it would be interesting to study the impact of misspecifying f on the estimation results. A sensitivity analysis was conducted for this purpose. The true parameters (including f) were the same as those of Table 1, but f was misspecified to be 0.01 or 0.1 for all of the five methods. The simulation results shown in Supporting Information Tables S16–S21 suggest that all methods were not affected by misspecifying the prevalence, except that the MSEs were slightly larger, with the prevalence being overspecified. This finding is consistent with those of Refs. [14, 20].
3.3
Significance test results
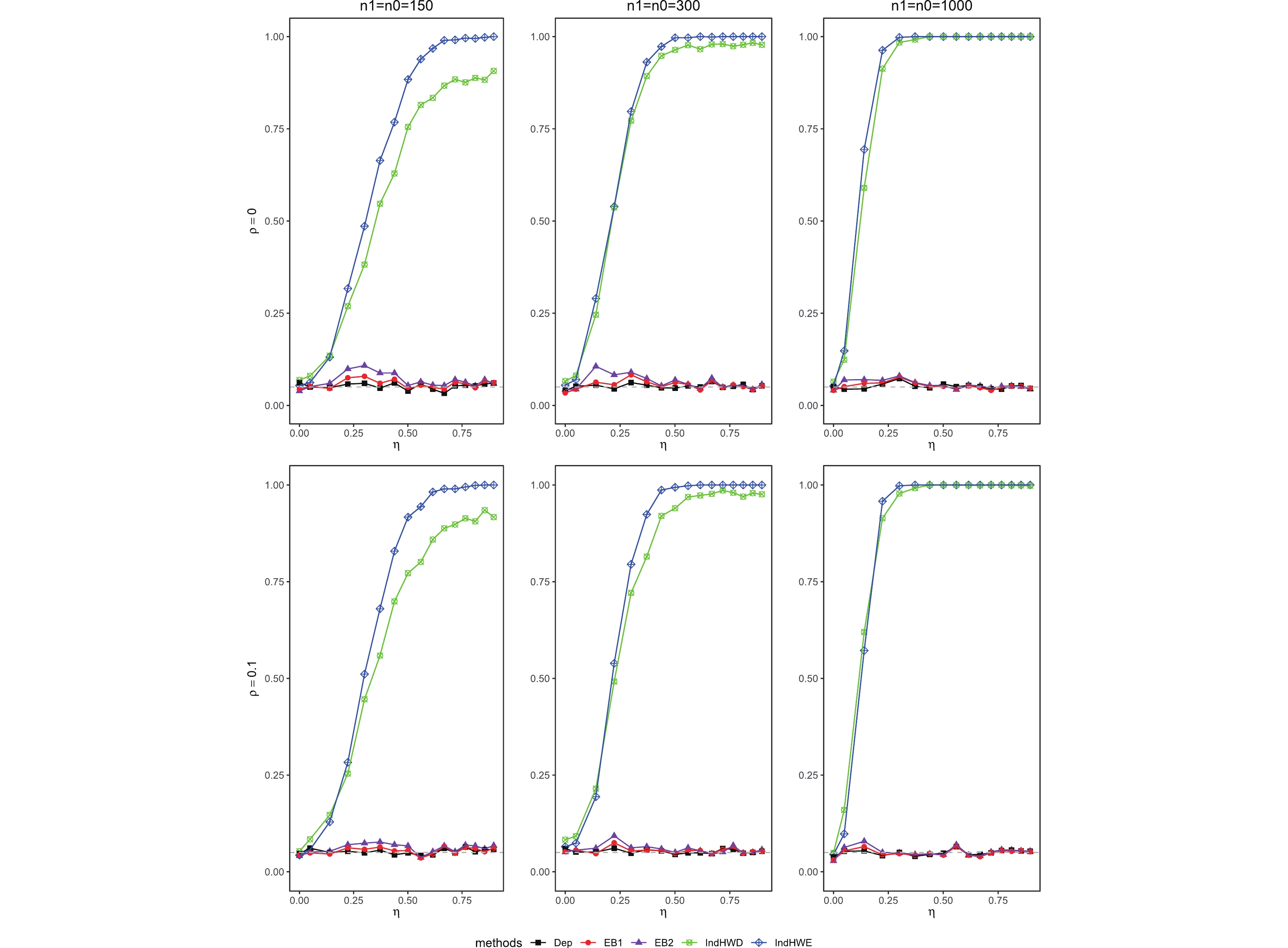
In this subsection, the significance test results (type-I error rates and powers at a significance level of 0.05) of the five considered methods are summarized for maternal gene-environment interactions in the simulation settings described in Section 3.1. Fig. 1 and Supporting Information Table S22 show type-I error rates for the significance tests of βmx1 for various η (0−log(2.5)), sample sizes (n1=n0=150, 300, and 1000), and ρ (0 for HWE and 0.1 for HWD). As expected, type-I error rates of IndHWE and IndHWD increase dramatically with η, because both methods rely on the independence assumption. In contrast, type-I error rates of both EB1 and EB2 are only slightly inflated (type I error rates: 4.4%−7.5% and 3.9%−9.9% for EB1 and EB2, respectively, n1=n0=150 and ρ=0), controlled around the nominal level even for large η, demonstrating the robustness of EB1 and EB2. Not surprisingly, Dep has well-controlled type-I error rates (4.4%−6.2%, n1=n0=150, and ρ=0).
Figure
1.
Type-I error rates for the significance tests of βmx1 with ρ=0 (HWE) or ρ=0.1 (HWD), various sample sizes (n1=n0=150, n1=n0=300, and n1=n0=1000), and various η values (0 through log(2.5)). The other parameters were fixed: βc=log(1.8), βm=log(1.3), βx1=βx2=−log(1.2), βcx1=βcx2=−log(1.5), and βmx2=log(1.2).
Figs. 2 and 3 show the powers for testing the Gm-X1 interaction for various βmx1 (from−log(1.5)tolog(1.5)), sample sizes (n1=n0=150, 300, and 1000), and ρ (0 for HWE and 0.1 for HWD). Because Type I error rates of IndHWE and IndHWD cannot be controlled when η>0, powers are not reported for IndHWE and IndHWD in such a situation. As expected, IndHWE is the most powerful among the five methods when the independence assumption holds (η=0), followed by IndHWD ranks the second. Evidently, EB1 and EB2 were more powerful than Dep in this situation. When the independence assumption does not hold, EB1 and EB2 can be either more or less powerful than Dep, depending on the sign of βmx1.
Figure
2.
Powers for the significance tests of maternal gene-environment interaction (βmx1) with HWE (ρ=0), various sample sizes (n1=n0=150, n1=n0=300, and n1=n0=1000), and various η values (0, log(1.2), and log(1.5)). The other parameters were fixed: βc=log(1.8), βm=log(1.3), βx1=βx2=−log(1.2), βcx1=βcx2=−log(1.5), and βmx2=log(1.2).
Figure
3.
Powers for the significance tests of maternal gene-environment interaction (βmx1) with HWD (ρ=0.1), various sample sizes (n1=n0=150, n1=n0=300, and n1=n0=1000), and various η values (0, log(1.2), and log(1.5)). The other parameters were fixed: βc=log(1.8), βm=log(1.3), βx1=βx2=−log(1.2), βcx1=βcx2=−log(1.5), and βmx2=log(1.2).
The diagrams of Type I errors and powers for the significance tests of the combined effect of maternal gene-environment interactions are shown in the Supporting Information Figures S1–S3, which have patterns similar to those of the Gm-X1 interaction effect.
4.
Application to the Danish National Birth Cohort study
Previous studies have shown that spontaneous preterm birth (SPTB) is the single largest cause of premature birth and is thought to be caused by a variety of factors, including infection or inflammation, stress, malnutrition, and genetic factors[5]. Using case-control mother-child pair data nested in the Danish National Birth Cohort (DNBC), we examined the association between SPTB and candidate SNPs by adjusting for maternal pre-pregnancy BMI (pp-BMI).
The DNBC is a cohort study that began in 1996. Extensive biomaterials and epidemiological data were collected from more than 10 0000 mothers and their children in Denmark. It has been well established that both fetal and maternal genotypes contribute to the risk of preterm birth, and genetic loci associated with SPTB have been identified through genetic association studies[21–24]. Furthermore, low maternal pp-BMI was believed to be associated with a high risk of SPTB[25]. In this study, 720 mothers gave birth prematurely (gestational days <259lt; 259$) and 906 term deliveries (280⩽ gestational days \leqslant 286) among 1626 eligible mother-child pairs.
The SNPs rs9939609 in genes FTO and rs2684811 in gene IGF1R have been identified as associated with BMI and SPTB, respectively[26, 23]. In the current study, we focused on the common SNPs located within 50kb of rs9939609 (eight SNPs in this region) and rs2684811 (24 SNPs in this region). All these 32 SNPs appear to be in HWE according to our analysis results by PLINK[27]; refer to Supporting Information Table S24 for the estimated MAFs and the p-values for testing HWE. Moreover, maternal pp-BMI can be assumed to be independent of the child genotype conditional on the maternal genotype since it is a maternal phenotype. Independent tests were also performed between pp-BMI and maternal genotype using the Kruskal-Wallis test. We applied the five methods mentioned in Section 3 to this dataset and adopted an additive mode of inheritance for each method. As the five methods are robust with respect to misspecification of disease prevalence according to our simulation results described in Section 3, we fixed the preterm birth prevalence at 7% because the prevalence of preterm birth in Denmark is approximately 5%–9% [28, 29] according to a survey of birth data in developed countries.
All the considered methods are expected to be nearly unbiased if G^{m} is independent of X , and EB1 and EB2 should be preferable to Dep in this situation. On the contrary, if G^{m} is strongly correlated to X , then IndHWE is generally biased, and EB1 and EB2 should be preferable to IndHWE and IndHWD. To verify this assertion, we studied the relationship between the shrinkage strengths of EB estimators and the association strength between X and G^m . First, a linear transformation was performed such that the transformed effects of Dep and IndHWE were 0 and 1, respectively. Then, the shrinkage strengths of the two EB estimators were measured using the regression coefficient between the transformed EB estimators and the p-value for testing the independence between X and G^m . As shown in Supporting Information Fig. S4, the desired shrinkage strength of EB1 is significant (regression coefficient = 0.398; 95% confidence interval = [0.130, 0.665]), which is effect-independent (refer to Supporting Information Appendix S5 for a rigorous proof). In contrast, the shrinkage strength of EB2 is significant only for the G^m - X interaction effect \beta_{mx} (regression {\rm coefficient} = 0.277; 95% confidence interval = [0.057, 0.497]). In the following, we describe the analysis results for two selected SNPs (rs16945088 and rs9941349) in detail, which are also summarized in Table 4. The results of the analysis of the other 30 candidate SNPs are summarized in Supporting Information Tables S25–S28.
Table
4.
Genetic effect estimates of two common SNPs in genes FTO and IGF1R (Danish National Birth Cohort).
SNPa
log(OR)
Dep
IndHWE
EB1
EB2
Estb
SEc
Pd
Estb
SEc
Pd
Estb
SEc
Pd
Estb
SEc
Pd
rs16945088
\beta_c
–0.151
0.140
0.283
–0.146
0.127
0.250
–0.146
0.127
0.249
–0.146
0.127
0.250
(0.636)
\beta_m
0.055
0.147
0.709
0.049
0.125
0.694
0.050
0.125
0.692
0.049
0.125
0.694
\beta_{cx}
0.045
0.118
0.702
0.044
0.115
0.703
0.044
0.116
0.703
0.044
0.115
0.703
\beta_{mx}
–0.081
0.160
0.613
–0.103
0.114
0.367
–0.102
0.116
0.382
–0.103
0.115
0.373
rs9941349
\beta_c
0.053
0.081
0.511
0.109
0.071
0.127
0.059
0.079
0.455
0.091
0.077
0.236
(0.039)
\beta_m
0.034
0.086
0.693
–0.031
0.074
0.676
0.027
0.083
0.747
–0.007
0.082
0.929
\beta_{cx}
–0.070
0.073
0.341
–0.071
0.073
0.327
–0.070
0.073
0.340
–0.071
0.073
0.327
\beta_{mx}
0.073
0.083
0.380
0.176
0.074
0.017
0.084
0.082
0.304
0.113
0.085
0.180
[Note] a Selected SNPs (the first one is BMI-related candidate SNP in gene FTO, and the second one is SPTB-related candidate SNP in gene IGF1R). Presented in parentheses are p-values for testing the independence between the SNPs and maternal pp-BMI using Kruskal-Wallis test. b Estimated effects. c Estimated standard errors. d Significance test p-values.
For the first SNP rs16945088, the maternal genotype is shown to be independent of maternal pp-BMI (\text{p-value} = 0.636). Consequently, the effect estimates of Dep and IndHWE are close to each other, IndHWE is generally more efficient than Dep in terms of standard errors, and the estimates of EB1 and EB2 are much closer to those of IndHWE than to Dep. For example, the \beta_{mx} estimates of Dep, IndHWE, EB1, and EB2 are -0.081 , -0.103 , -0.102 , and -0.103 , respectively, indicating that the estimates of EB1 and EB2 are much closer to that of IndHWE than those of Dep. In addition, the standard errors of the \beta_{mx} estimates by IndHWE, EB1, and EB2 are 0.114, 0.116, and 0.115, respectively, compared to 0.160 by Dep, indicating that the former three methods are more efficient than Dep. For the second SNP rs9941349, maternal genotype is significantly associated with pp-BMI (p-value = 0.039). As a result, the estimates of EB1 are closer to those of Dep compared with IndHWE, whereas the estimates of EB2 are closer to those of IndHWE compared with Dep except that the \beta_{mx} estimate by EB2 lies in the middle of the \beta_{mx} estimates of Dep and IndHWE. For example, the \beta_{mx} estimates of Dep, EB1, and EB2 are 0.073, 0.084, and 0.113, respectively, compared to 0.176 by IndHWE. Furthermore, the G^m - X interaction is not significant for Dep, EB1, and EB2 (\text{p-values} > 0.3), whereas it is significant for IndHWE (\text{p-value}= 0.017). This suggests that the seemingly significant result of IndHWE might be a false positive finding.
5.
Conclusions
In this study, two EB-type estimators (EB1 and EB2) are proposed by appropriately weighting an independent model estimator and a dependent-model estimator, which can adaptively balance robustness and efficiency. The independent-model estimator is an efficient estimator that improves efficiency by imposing HWE and G^m - X independence assumptions in the model, but it could produce a serious estimation bias if at least one of the assumptions is violated. In contrast, the dependent-model estimator Dep is a robust estimator that does not require the above two assumptions but might not be efficient. The EB-type estimators EB1 and EB2 developed in this study adaptively utilize the HWE assumption and the independence assumption between maternal genotype and maternal environmental risk factors, which appropriately balance robustness and efficiency. As in Dep, EB1 and EB2 fully utilize Mendelian inheritance and the conditional independence assumption between maternal environmental risk factors and children’s genotype given maternal genotype.
These properties were verified by our simulation results in various situations. Specifically, if both HWE and G^m - X independence assumptions hold, EB1 and EB2 are more efficient than Dep, although they are less efficient than IndHWE. In contrast, IndHWE could be seriously biased if HWE or G^m - X independence assumptions are violated, while EB1 and EB2 still perform well. If the sample size is small, EB1 is more robust than EB2, whereas EB2 appears more suitable for moderate or large samples in terms of estimation efficiency. The desired properties of EB1 and EB2 are also demonstrated using a real-data application. In practice, it is difficult to obtain prior information regarding the relationship between maternal genotype and environmental risk factors. In this situation, EB1 and EB2 serve as robust alternatives to IndHWE and Dep.
Our novel method has been designed to analyze common genetic variants (MAF\geqslant 5\%) and the low-dimensional covariate X . In practice, there could be many mother-related characteristics, such as age, pre-pregnancy BMI, and smoking. The dimension of the parameters could be at least three times that of X when studying gene-environment interactions in case-control mother-child pair data. With high-dimensional parameters, we can adopt any regularization method to select associated covariates. However, increasing evidence shows that rare genetic variants (MAF< 1\%lt; 1\%$) play an important role in complex diseases and traits[30–32]. Recently, some methods have been developed to analyze family based multilocus rare variants[33–36]. However, these methods are not based on retrospective likelihood functions; therefore, they cannot fully utilize family information, such as Mendelian inheritance and gene-environment independence. Further investigation is thus needed to extend our method to analyze multilocus rare variants under a case-control mother-child pair design.
Data availability statement
The DNBC data are available at the dbGAP website (study accession: phs000103.v1.p1).
Supporting information
The supporting information for this article is available online at https://doi.org/10.52396/JUSTC-2022-0007. It includes Appendices S1–S5, Figures S1–S4, and Tables S1–S28.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (11771096, 12171451, and 72091212). Funding support for the GWAS of Prematurity and Its Complications study was provided by the NIH Genes, Environment, and Health Initiative [GEI] (U01HG004423). The GWAS of Prematurity and its Complications study is one of the genome-wide association studies funded as part of the Gene Environment Association Studies (GENEVA) under GEI. Assistance with phenotype harmonization and genotype cleaning, as well as general study coordination, was provided by the GENEVA Coordinating Center (U01HG004446). The National Center for Biotechnology Information provided assistance with data cleaning. Funding support for genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research, was provided by the NIH GEI (U01HG004438) and the NIH contract “High-throughput Genotyping for Studying the Genetic Contributions to Human Disease” (HHSN268200782096C).
Conflict of interest
The authors declare that they have no conflict of interest.
Conflict of Interest
The authors declare that they have no conflict of interest.
Retrospective likelihood method is employed to improve statistical efficiency by fullyutilizing available information.
An efficient estimator and a robust estimator are combined to construct twonovel empirical Bayes-type estimators using empirical the Bayes method.
We establish the asymptotic properties of the proposed estimators.
Goddard K A, Tromp G, Romero R, et al. Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes. Human Heredity,2007, 63 (1): 1–16. DOI: 10.1159/000097926
[2]
Kanayama N, Takahashi K, Matsuura T, et al. Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Molecular Human Reproduction,2002, 8 (12): 1129–1135. DOI: 10.1093/molehr/8.12.1129
[3]
Saftlas A F, Beydoun H, Triche E. Immunogenetic determinants of preeclampsia and related pregnancy disorders: A systematic review. Obstetrics and Gynecology,2005, 106 (1): 162–172. DOI: 10.1097/01.AOG.0000167389.97019.37
[4]
Wangler M F, Chang A S, Moley K H, et al. Factors associated with preterm delivery in mothers of children with Beckwith-Wiedemann syndrome: A case cohort study from the BWS registry. American Journal of Medical Genetics Part A,2005, 134 (2): 187–191. DOI: 10.1002/ajmg.a.30595
[5]
Goldenberg R L, Culhane J F, Iams J D, et al. Epidemiology and causes of preterm birth. The Lancet,2008, 371 (9606): 75–84. DOI: 10.1016/S0140-6736(08)60074-4
[6]
Zhang G, Feenstra B, Bacelis J, et al. Genetic associations with gestational duration and spontaneous preterm birth. The New England Journal of Medicine,2017, 377 (12): 1156–1167. DOI: 10.1056/NEJMoa1612665
[7]
Hong X, Hao K, Ji H, et al. Genome-wide approach identifies a novel gene-maternal pre-pregnancy BMI interaction on preterm birth. Nature Communications,2017, 8 (1): 15608. DOI: 10.1038/ncomms15608
[8]
Chen J, Zheng H, Wilson M L. Likelihood ratio tests for maternal and fetal genetic effects on obstetric complications. Genetic Epidemiology,2009, 33 (6): 526–538. DOI: 10.1002/gepi.20405
[9]
Fu W, Li M, Sun K, et al. Testing maternal-fetal genotype incompatibility with mother-offspring pair data. Journal of Proteomics and Genomics Research,2013, 1 (2): 40–56. DOI: 10.14302/issn.2326-0793.jpgr-12-160
[10]
Chen J, Lin D, Hochner H. Semiparametric maximum likelihood methods for analyzing genetic and environmental effects with case-control mother-child pair data. Biometrics,2012, 68 (3): 869–877. DOI: 10.1111/j.1541-0420.2011.01728.x
[11]
Lin D, Weinberg C R, Feng R, et al. A multi-locus likelihood method for assessing parent-of-origin effects using case-control mother-child pairs. Genetic Epidemiology,2013, 37 (2): 152–162. DOI: 10.1002/gepi.21700
[12]
Prentice R L, Pyke R. Logistic disease incidence models and case-control studies. Biometrika,1979, 66 (3): 403–411. DOI: 10.1093/biomet/66.3.403
[13]
Shi M, Umbach D M, Vermeulen S H, et al. Making the most of case-mother/control-mother studies. American Journal of Epidemiology,2008, 168 (5): 541–547. DOI: 10.1093/aje/kwn149
[14]
Zhang H, Mukherjee B, Arthur V, et al. An efficient and computationally robust statistical method for analyzing case-control mother-offspring pair genetic association studies. Annals of Applied Statics,2020, 14 (2): 560–584. DOI: 10.1214/19-AOAS1298
[15]
Chen Y H, Chatterjee N, Carroll R J. Shrinkage estimators for robust and efficient inference in haplotype-based case-control studies. Journal of the American Statistical Association,2009, 104 (485): 220–233. DOI: 10.1198/jasa.2009.0104
[16]
Owen A B. Empirical Likelihood. New York: Chapman and Hall/ CRC, 2001.
[17]
Zhang H, Chatterjee N, Rader D, et al. Adjustment of nonconfounding covariates in case-control genetic association studies. Annals of Applied Statistics,2018, 12 (1): 200–221. DOI: 10.1214/17-AOAS1065
[18]
Casella G, Berger R L. Statistical Inference. 2nd edition. Boston, MA: Cengage Learning, 2001.
[19]
Mukherjee B, Chatterjee N. Exploiting gene-environment independence for analysis of case-control studies: An empirical Bayes-type shrinkage estimator to trade-off between bias and efficiency. Biometrics,2008, 64 (3): 685–694. DOI: 10.1111/j.1541-0420.2007.00953.x
[20]
Zhang K, Zhang H, Hochner H, et al. Covariate adjusted inference of parent-of-origin effects using case-control mother-child paired multilocus genotype data. Genetic Epidemiology,2021, 45 (8): 830–847. DOI: 10.1002/gepi.22428
[21]
Engel S A M, Erichsen H C, Savitz D A, et al. Risk of spontaneous preterm birth is associated with common proinflammatory cytokine polymorphisms. Epidemiology,2005, 16 (4): 469–477. DOI: 10.1097/01.ede.0000164539.09250.31
[22]
Frey H A, Stout M J, Pearson L N, et al. Genetic variation associated with preterm birth in African-American women. American Journal of Obstetrics and Gynecology,2016, 215 (2): 235.e1–235.e8. DOI: 10.1016/j.ajog.2016.03.008
[23]
Haataja R, Karjalainen M K, Luukkonen A, et al. Mapping a new spontaneous preterm birth susceptibility gene, IGF1R, using linkage, haplotype sharing, and association analysis. PLoS Genetics,2011, 7 (2): e1001293. DOI: 10.1371/journal.pgen.1001293
[24]
Menon R, Velez D R, Simhan H, et al. Multilocus interactions at maternal tumor necrosis factor-α, tumor necrosis factor receptors, interleukin-6 and interleukin-6 receptor genes predict spontaneous preterm labor in European-American women. American Journal of Obstetrics and Gynecology,2006, 194 (6): 1616–1624. DOI: 10.1016/j.ajog.2006.03.059
[25]
Hendler I, Goldenberg R L, Mercer B M, et al. The preterm prediction study: Association between maternal body mass index and spontaneous and indicated preterm birth. American Journal of Obstetrics and Gynecology,2005, 192 (3): 882–886. DOI: 10.1016/j.ajog.2004.09.021
[26]
Frayling T M, Timpson N J, Weedon M N, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science,2007, 316 (5826): 889–894. DOI: 10.1126/science.1141634
[27]
Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics,2007, 81 (3): 559–575. DOI: 10.1086/519795
[28]
Hamilton B E, Martin J A, Ventura S J. Births: Preliminary data for 2005. National Vital Statistics Reports,2006, 55 (11): 1–18.
[29]
Slattery M M, Morrison J J. Preterm delivery. The Lancet,2002, 360 (9344): 1489–1497. DOI: 10.1016/S0140-6736(02)11476-0
[30]
Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genetics,2008, 40 (6): 695–701. DOI: 10.1038/ng.f.136
[31]
Lee S, Abecasis G R, Boehnke M, et al. Rare-variant association analysis: Study designs and statistical tests. American Journal of Human Genetics,2014, 95 (1): 5–23. DOI: 10.1016/j.ajhg.2014.06.009
[32]
Schork N J, Murray S S, Frazer K A, et al. Common vs. rare allele hypotheses for complex diseases. Current Opinion in Genetics and Development,2009, 19 (3): 212–219. DOI: 10.1016/j.gde.2009.04.010
[33]
Ionita-Laza I, Lee S, Makarov V, et al. Family-based association tests for sequence data, and comparisons with population-based association tests. European Journal of Human Genetics,2013, 21 (10): 1158–1162. DOI: 10.1038/ejhg.2012.308
[34]
Jiang D, McPeek M S. Robust rare variant association testing for quantitative traits in samples with related individuals. Genetic Epidemiology,2014, 38 (1): 10–20. DOI: 10.1002/gepi.21775
[35]
Wang X, Lee S, Zhu X, et al. GEE-based SNP set association test for continuous and discrete traits in family-based association studies. Genetic Epidemiology,2013, 37 (8): 778–786. DOI: 10.1002/gepi.21763
[36]
Wang X, Zhang Z, Morris N, et al. Rare variant association test in family-based sequencing studies. Briefings in Bioinformatics,2016, 18 (6): 954–961. DOI: 10.1093/bib/bbw083
Figure
1.
Type-I error rates for the significance tests of \beta_{mx_1} with \rho=0 (HWE) or \rho=0.1 (HWD), various sample sizes ( n_1=n_0=150 , n_1=n_0=300 , and n_1=n_0=1000 ), and various \eta values ( 0 through \log(2.5) ). The other parameters were fixed: \beta_{c}=\text{log}(1.8) , \beta_{m}=\text{log}(1.3) , \beta_{x_1}=\beta_{x_2}=-\text{log}(1.2) , \beta_{cx_1}=\beta_{cx_2}=-\text{log}(1.5) , and \beta_{mx_2}=\text{log}(1.2) .
Figure
2.
Powers for the significance tests of maternal gene-environment interaction ( \beta_{mx_1} ) with HWE ( \rho=0 ), various sample sizes ( n_1=n_0=150 , n_1=n_0=300 , and n_1=n_0=1000 ), and various \eta values ( 0 , \log(1.2) , and \log(1.5) ). The other parameters were fixed: \beta_{c}=\text{log}(1.8) , \beta_{m}=\text{log}(1.3) , \beta_{x_1}=\beta_{x_2}=-\text{log}(1.2) , \beta_{cx_1}=\beta_{cx_2}=-\text{log}(1.5) , and \beta_{mx_2}=\text{log}(1.2) .
Figure
3.
Powers for the significance tests of maternal gene-environment interaction ( \beta_{mx_1} ) with HWD ( \rho=0.1 ), various sample sizes ( n_1=n_0=150 , n_1=n_0=300 , and n_1=n_0=1000 ), and various \eta values ( 0 , \log(1.2) , and \log(1.5) ). The other parameters were fixed: \beta_{c}=\text{log}(1.8) , \beta_{m}=\text{log}(1.3) , \beta_{x_1}=\beta_{x_2}=-\text{log}(1.2) , \beta_{cx_1}=\beta_{cx_2}=-\text{log}(1.5) , and \beta_{mx_2}=\text{log}(1.2) .
References
[1]
Goddard K A, Tromp G, Romero R, et al. Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes. Human Heredity,2007, 63 (1): 1–16. DOI: 10.1159/000097926
[2]
Kanayama N, Takahashi K, Matsuura T, et al. Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Molecular Human Reproduction,2002, 8 (12): 1129–1135. DOI: 10.1093/molehr/8.12.1129
[3]
Saftlas A F, Beydoun H, Triche E. Immunogenetic determinants of preeclampsia and related pregnancy disorders: A systematic review. Obstetrics and Gynecology,2005, 106 (1): 162–172. DOI: 10.1097/01.AOG.0000167389.97019.37
[4]
Wangler M F, Chang A S, Moley K H, et al. Factors associated with preterm delivery in mothers of children with Beckwith-Wiedemann syndrome: A case cohort study from the BWS registry. American Journal of Medical Genetics Part A,2005, 134 (2): 187–191. DOI: 10.1002/ajmg.a.30595
[5]
Goldenberg R L, Culhane J F, Iams J D, et al. Epidemiology and causes of preterm birth. The Lancet,2008, 371 (9606): 75–84. DOI: 10.1016/S0140-6736(08)60074-4
[6]
Zhang G, Feenstra B, Bacelis J, et al. Genetic associations with gestational duration and spontaneous preterm birth. The New England Journal of Medicine,2017, 377 (12): 1156–1167. DOI: 10.1056/NEJMoa1612665
[7]
Hong X, Hao K, Ji H, et al. Genome-wide approach identifies a novel gene-maternal pre-pregnancy BMI interaction on preterm birth. Nature Communications,2017, 8 (1): 15608. DOI: 10.1038/ncomms15608
[8]
Chen J, Zheng H, Wilson M L. Likelihood ratio tests for maternal and fetal genetic effects on obstetric complications. Genetic Epidemiology,2009, 33 (6): 526–538. DOI: 10.1002/gepi.20405
[9]
Fu W, Li M, Sun K, et al. Testing maternal-fetal genotype incompatibility with mother-offspring pair data. Journal of Proteomics and Genomics Research,2013, 1 (2): 40–56. DOI: 10.14302/issn.2326-0793.jpgr-12-160
[10]
Chen J, Lin D, Hochner H. Semiparametric maximum likelihood methods for analyzing genetic and environmental effects with case-control mother-child pair data. Biometrics,2012, 68 (3): 869–877. DOI: 10.1111/j.1541-0420.2011.01728.x
[11]
Lin D, Weinberg C R, Feng R, et al. A multi-locus likelihood method for assessing parent-of-origin effects using case-control mother-child pairs. Genetic Epidemiology,2013, 37 (2): 152–162. DOI: 10.1002/gepi.21700
[12]
Prentice R L, Pyke R. Logistic disease incidence models and case-control studies. Biometrika,1979, 66 (3): 403–411. DOI: 10.1093/biomet/66.3.403
[13]
Shi M, Umbach D M, Vermeulen S H, et al. Making the most of case-mother/control-mother studies. American Journal of Epidemiology,2008, 168 (5): 541–547. DOI: 10.1093/aje/kwn149
[14]
Zhang H, Mukherjee B, Arthur V, et al. An efficient and computationally robust statistical method for analyzing case-control mother-offspring pair genetic association studies. Annals of Applied Statics,2020, 14 (2): 560–584. DOI: 10.1214/19-AOAS1298
[15]
Chen Y H, Chatterjee N, Carroll R J. Shrinkage estimators for robust and efficient inference in haplotype-based case-control studies. Journal of the American Statistical Association,2009, 104 (485): 220–233. DOI: 10.1198/jasa.2009.0104
[16]
Owen A B. Empirical Likelihood. New York: Chapman and Hall/ CRC, 2001.
[17]
Zhang H, Chatterjee N, Rader D, et al. Adjustment of nonconfounding covariates in case-control genetic association studies. Annals of Applied Statistics,2018, 12 (1): 200–221. DOI: 10.1214/17-AOAS1065
[18]
Casella G, Berger R L. Statistical Inference. 2nd edition. Boston, MA: Cengage Learning, 2001.
[19]
Mukherjee B, Chatterjee N. Exploiting gene-environment independence for analysis of case-control studies: An empirical Bayes-type shrinkage estimator to trade-off between bias and efficiency. Biometrics,2008, 64 (3): 685–694. DOI: 10.1111/j.1541-0420.2007.00953.x
[20]
Zhang K, Zhang H, Hochner H, et al. Covariate adjusted inference of parent-of-origin effects using case-control mother-child paired multilocus genotype data. Genetic Epidemiology,2021, 45 (8): 830–847. DOI: 10.1002/gepi.22428
[21]
Engel S A M, Erichsen H C, Savitz D A, et al. Risk of spontaneous preterm birth is associated with common proinflammatory cytokine polymorphisms. Epidemiology,2005, 16 (4): 469–477. DOI: 10.1097/01.ede.0000164539.09250.31
[22]
Frey H A, Stout M J, Pearson L N, et al. Genetic variation associated with preterm birth in African-American women. American Journal of Obstetrics and Gynecology,2016, 215 (2): 235.e1–235.e8. DOI: 10.1016/j.ajog.2016.03.008
[23]
Haataja R, Karjalainen M K, Luukkonen A, et al. Mapping a new spontaneous preterm birth susceptibility gene, IGF1R, using linkage, haplotype sharing, and association analysis. PLoS Genetics,2011, 7 (2): e1001293. DOI: 10.1371/journal.pgen.1001293
[24]
Menon R, Velez D R, Simhan H, et al. Multilocus interactions at maternal tumor necrosis factor-α, tumor necrosis factor receptors, interleukin-6 and interleukin-6 receptor genes predict spontaneous preterm labor in European-American women. American Journal of Obstetrics and Gynecology,2006, 194 (6): 1616–1624. DOI: 10.1016/j.ajog.2006.03.059
[25]
Hendler I, Goldenberg R L, Mercer B M, et al. The preterm prediction study: Association between maternal body mass index and spontaneous and indicated preterm birth. American Journal of Obstetrics and Gynecology,2005, 192 (3): 882–886. DOI: 10.1016/j.ajog.2004.09.021
[26]
Frayling T M, Timpson N J, Weedon M N, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science,2007, 316 (5826): 889–894. DOI: 10.1126/science.1141634
[27]
Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics,2007, 81 (3): 559–575. DOI: 10.1086/519795
[28]
Hamilton B E, Martin J A, Ventura S J. Births: Preliminary data for 2005. National Vital Statistics Reports,2006, 55 (11): 1–18.
[29]
Slattery M M, Morrison J J. Preterm delivery. The Lancet,2002, 360 (9344): 1489–1497. DOI: 10.1016/S0140-6736(02)11476-0
[30]
Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genetics,2008, 40 (6): 695–701. DOI: 10.1038/ng.f.136
[31]
Lee S, Abecasis G R, Boehnke M, et al. Rare-variant association analysis: Study designs and statistical tests. American Journal of Human Genetics,2014, 95 (1): 5–23. DOI: 10.1016/j.ajhg.2014.06.009
[32]
Schork N J, Murray S S, Frazer K A, et al. Common vs. rare allele hypotheses for complex diseases. Current Opinion in Genetics and Development,2009, 19 (3): 212–219. DOI: 10.1016/j.gde.2009.04.010
[33]
Ionita-Laza I, Lee S, Makarov V, et al. Family-based association tests for sequence data, and comparisons with population-based association tests. European Journal of Human Genetics,2013, 21 (10): 1158–1162. DOI: 10.1038/ejhg.2012.308
[34]
Jiang D, McPeek M S. Robust rare variant association testing for quantitative traits in samples with related individuals. Genetic Epidemiology,2014, 38 (1): 10–20. DOI: 10.1002/gepi.21775
[35]
Wang X, Lee S, Zhu X, et al. GEE-based SNP set association test for continuous and discrete traits in family-based association studies. Genetic Epidemiology,2013, 37 (8): 778–786. DOI: 10.1002/gepi.21763
[36]
Wang X, Zhang Z, Morris N, et al. Rare variant association test in family-based sequencing studies. Briefings in Bioinformatics,2016, 18 (6): 954–961. DOI: 10.1093/bib/bbw083
Table
1.
MSE and bias (in parenthesis) of various estimators when case and control pairs are equal and both the independence assumption ( \eta=0 ) and HWE ( \rho=0 ) hold.
Table
2.
MSE and bias (in parenthesis) of various estimators when case and control pairs are equal and the independence assumption is violated (\eta={\rm log}(1.5)) but HWE holds ( \rho=0 ).
Table
3.
MSE and bias (in parenthesis) of various estimators when case and control pairs are equal and the independence assumption is violated (\eta={\rm log}(2.5)) but HWE holds ( \rho=0 ).
Table
4.
Genetic effect estimates of two common SNPs in genes FTO and IGF1R (Danish National Birth Cohort).
SNPa
log(OR)
Dep
IndHWE
EB1
EB2
Estb
SEc
Pd
Estb
SEc
Pd
Estb
SEc
Pd
Estb
SEc
Pd
rs16945088
\beta_c
–0.151
0.140
0.283
–0.146
0.127
0.250
–0.146
0.127
0.249
–0.146
0.127
0.250
(0.636)
\beta_m
0.055
0.147
0.709
0.049
0.125
0.694
0.050
0.125
0.692
0.049
0.125
0.694
\beta_{cx}
0.045
0.118
0.702
0.044
0.115
0.703
0.044
0.116
0.703
0.044
0.115
0.703
\beta_{mx}
–0.081
0.160
0.613
–0.103
0.114
0.367
–0.102
0.116
0.382
–0.103
0.115
0.373
rs9941349
\beta_c
0.053
0.081
0.511
0.109
0.071
0.127
0.059
0.079
0.455
0.091
0.077
0.236
(0.039)
\beta_m
0.034
0.086
0.693
–0.031
0.074
0.676
0.027
0.083
0.747
–0.007
0.082
0.929
\beta_{cx}
–0.070
0.073
0.341
–0.071
0.073
0.327
–0.070
0.073
0.340
–0.071
0.073
0.327
\beta_{mx}
0.073
0.083
0.380
0.176
0.074
0.017
0.084
0.082
0.304
0.113
0.085
0.180
[Note] a Selected SNPs (the first one is BMI-related candidate SNP in gene FTO, and the second one is SPTB-related candidate SNP in gene IGF1R). Presented in parentheses are p-values for testing the independence between the SNPs and maternal pp-BMI using Kruskal-Wallis test. b Estimated effects. c Estimated standard errors. d Significance test p-values.


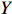

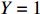
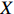
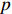
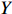
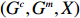
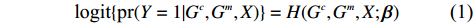
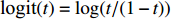
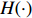
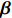
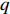
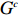
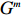
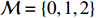
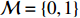
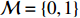
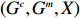
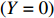
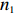
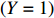
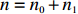
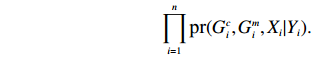
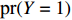
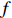
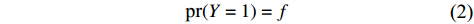
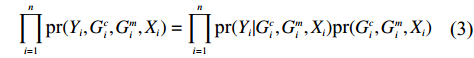
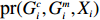
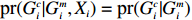
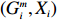
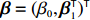
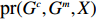
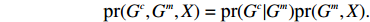
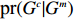
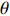
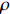
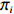
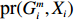
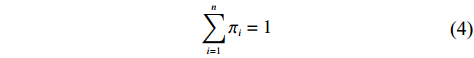
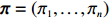
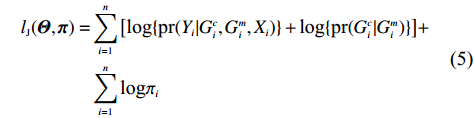
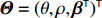
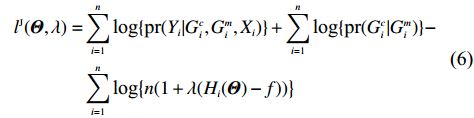
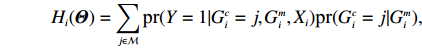
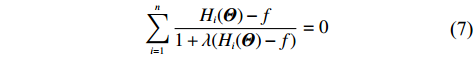
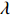
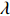
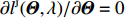
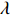
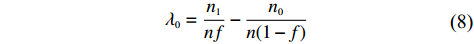
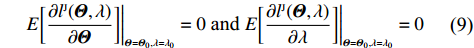
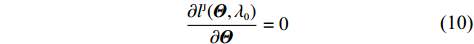
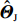
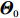
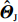
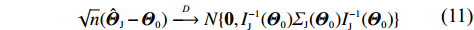
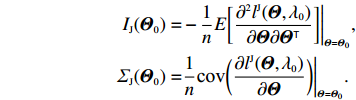
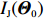
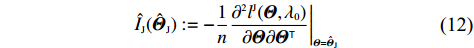
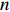
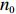
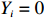
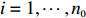
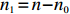
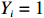
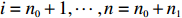
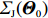
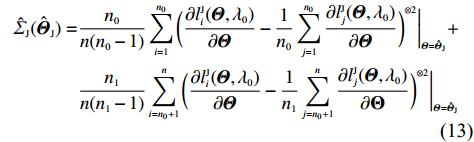

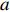
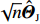
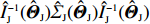
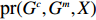
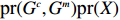
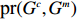
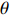
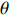
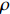
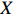
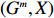
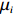
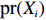
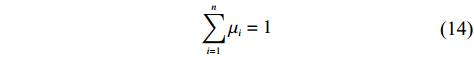
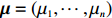
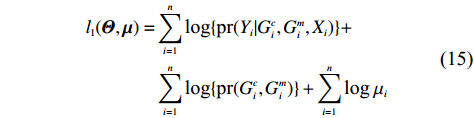
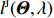
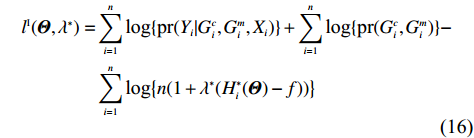
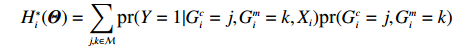
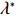
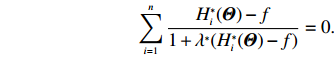

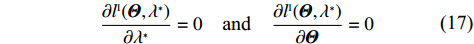


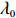
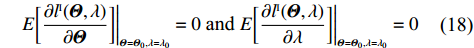

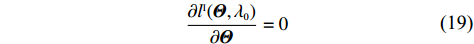

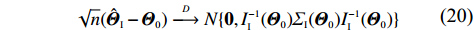




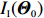
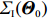


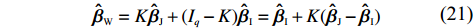





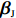

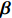







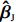






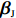
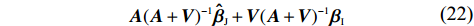
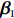
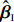






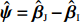


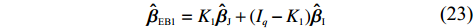

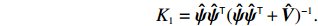
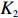
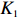

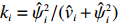
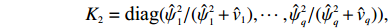





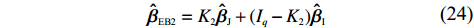
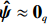
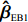
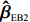

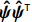









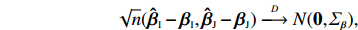
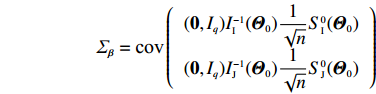








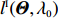


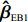







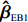



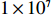




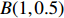







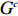
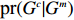



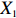


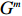






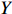



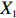


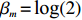

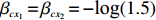

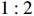






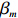



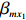
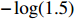
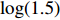



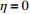

 DownLoad:
DownLoad: